



よくあるご質問
新規申請
- Q
- 委員会の審査結果が「継続審査」となったのですが、何をすればいいですか?
- A
- 委員会からの指示事項に従い、該当書類の修正・変更をし、統一書式3_変更審査依頼書とともに審査結果通知書受領後1週間程度でご提出ください。
- Q
- 委員会の審査結果が「継続審査」となったのですが、委員会への出席が必要ですか?
- A
- 基本的に出席の必要はありません。
委員会からの指示と大幅に異なる変更の場合、委員からのさらなる質問が予想される場合、個別に出席をお願いする場合があります。
変更申請(CRB承認後)
- Q
- 変更申請書類が委員会で審議された後は何をすればいいですか?
- A
- CRB審議後、 1~2週間程度でCRB事務局から審査結果通知書をメールでお送りします。その後まもなく、臨床研究の実施に関する決定通知書(管理者許可)をCRB事務局からメールでお送りしますので、そちらをお待ちください。変更内容が承認され、実施計画の変更があった場合にはjRCTに一時保存した内容に審査結果通知書を添付してご登録ください。
- Q
- 委員会で承認された変更後の書類(説明同意文書など)はいつから使用できますか?
- A
- CRB承認後、すぐには使用できません。CRB承認後、実施の管理者の許可が必要となります。また、jRCTに公開されていることも必要となります。具体的な変更文書の使用開始については、研究計画書に記載されている研究調整事務局にご確認ください。
- Q
- jRCTの操作方法がよくわかりません。
- A
- 臨床研究等提出・公開システム操作マニュアル【登録者編】の「業務を想定した操作方法」に詳細な記載がありますので、ご確認ください。
軽微変更
- Q
- 軽微変更とはなんですか?
- A
- 軽微変更とは、jRCTに登録した実施計画の変更においてCRB審議不要となる場合を指します。実施計画のみが対象のため、研究計画書や同意説明文書に軽微変更を適用することはできません。
次に掲げる変更が生じる場合には、軽微な変更を行ってください。
実施計画上の記載欄の
大項目・中項目該当する変更箇所 備考 1. 実施体制に関する事項及び施設の構造設備に関する事項 (2)(3)(4) 以下の氏名、連絡先又は所属する機関の名称の変更 - 研究責任(代表)医師の連絡先
- 統計解析担当機関・責任者
- 研究代表(責任)医師以外の研究を統括する者
- 婚姻などの氏名の変更は該当します。
- 当該者又は当該者の所属する機関の変更を伴う場合は該当しません。
(2)(4) 実施医療機関の管理者の氏名の変更 (2)(4) 医療機関の管理者の許可の有無に伴う変更 3. 特定臨床研究の実施状況の確認に関する事項 (2) - 症例登録開始予定日
- 第1症例登録日
- 進捗状況
主たる評価項目に係る研究結果の変更を伴う場合は該当しません。 6. 審査意見業務を行う認定臨床研究審査委員会の名称等 (2) 認定臨床研究審査委員会の名称又は連絡先の変更 - 委員会の名称
- 住所
- 電話番号
- 電子メールアドレス
認定臨床研究審査委員会(本体)の変更を伴うものは該当しません。 詳細は臨床研究等提出・公開システム操作マニュアル【登録者編】をご参照ください。
判断に迷う場合には、CRB事務局(prc-jim(アットマーク)chiba-u.jp)までお問い合わせください。
- Q
- 軽微変更の手続きは何をどこですればいいのですか?
- A
- jRCTの「軽微変更」のタブから、変更内容をご入力ください。詳細な操作方法は臨床研究等提出・公開システム操作マニュアル【登録者編】をご参照ください。ご登録後、実施計画(様式第一)と実施計画変更事項届書(様式第二)をCRB事務局 (prc-jim(アットマーク)chiba-u.jp)までご提出ください。
- Q
- 軽微変更の手続きはいつまでにすればいいのですか?
- A
- 当該変更後、10日以内にjRCTの登録とCRB事務局(prc-jim(アットマーク)chiba-u.jp)への報告をお願いします。
- Q
- 軽微変更後は、いつからその変更を適用できますか?
- A
- 軽微変更に該当する場合には、当該変更が生じたときから、その変更の適用が可能です。ただし、上述のとおり、その変更から10日以内にjRCTの登録とCRB事務局(prc-jim(アットマーク)chiba-u.jp)への報告が必要となります。
届出外変更
- Q
- 届出外変更とは何ですか?
- A
- 届出外変更とは、「実施計画」に記載がなく、jRCTには公開されている情報に対する変更を指します。届出外変更では、認定臨床研究審査委員会での審査・報告が不要です。但し、jRCT上での変更は必要となりますので、変更後は必ず登録を行ってください。
届出外変更には以下の変更が該当します。
- データマネジメント担当機関/データマネジメント担当責任者
- モニタリング担当機関/モニタリング担当責任者
- 監査担当機関/監査担当責任者
- 研究・開発計画支援担当機関/研究・開発計画支援担当者
- 調整・管理実務担当機関/調整・管理実務担当者
詳細は臨床研究等提出・公開システム操作マニュアル【登録者編】をご参照ください。判断に迷う場合には、CRB事務局 (prc-jim(アットマーク)chiba-u.jp) までお問い合わせください。
- Q
- 届出外変更では何をすればいいのですか?
- A
- 通常の変更申請と同様に、jRCTの「届出外変更」のタブから変更内容を登録します。詳細な操作方法は臨床研究等提出・公開システム操作マニュアル【登録者編】をご参照ください。届出外変更の手続きは以上ですが、届出外変更により、研究計画書の実施体制の項目などに変更が必要となる場合には、別途、研究計画書の変更申請を行ってください。
- Q
- 軽微変更とは何が違うのですか?
- A
- どちらもCRB審議が不要となる変更ですが、軽微変更と届出外変更では適用できる変更事項が異なります。また、軽微変更ではCRB事務局への変更後の書類提出が必要となりますが、届出外変更では不要となる点で異なります。
事前確認不要事項に関する変更
- Q
- 事前確認不要事項に関する変更とは何ですか?
- A
- 研究計画書や同意説明文書などの変更のうち、研究の実施に影響を与えない実施体制の変更等を指します。事前確認不要事項に関する変更は、CRBによる委員会審査ではなく簡易審査が適用されます。
千葉大学のCRB手順書では、以下の変更が事前確認不要事項に該当すると定められています。該当する変更内容 注意点 ①臨床研究従事者の職名変更 担当者の所属機関の変更を伴う場合は該当しません。 ②誤記等の事務的な変更 研究内容の変更を伴わないことが明らかである誤記の修正、記載整備又は別に定める手順書等の変更が審査され承認されたことに伴う実施体制の記載整備等に限ります。 判断に迷う場合には、CRB事務局(prc-jim(アットマーク)chiba-u.jp)までお問い合わせください。
- Q
- 事前確認不要事項に関する変更では何をすればいいのですか?
- A
- 変更資料をCRB事務局(prc-jim(アットマーク)chiba-u.jp)までご提出ください。その際に、通常の変更申請と同様に、変更審査依頼書は必要となります。
- Q
- 軽微変更や届出外変更と事前確認不要事項は何が違うのですか?
- A
- 軽微変更や届出外変更は、CRB審査は不要となります。事前確認不要事項に関する変更はあくまでCRB審査が簡易に行われる点が異なります。
疾病等報告
- Q
- 疾病等とは何ですか?
- A
- 臨床研究を実施することが原因(副作用など)で発生した疾病や障害、死亡、感染症を指します。疾病等が発生した場合、責任医師は厚生労働大臣やCRBへ報告することが必要となる場合があります。詳細は2つ下以降のQ.をご覧ください。
- Q
- 不具合とは何ですか?
- A
- 試験機器について、破損、作動不良等広く品質、安全性、性能等に関する試験機器の具合が良くないことを言います。設計、交付、保管、使用のいずれの段階によるものであるかを問いません。
- Q
- CRBへの疾病等報告はどのようなときに必要になりますか?
- A
- 疾病等が発生した場合、以下の期限でCRBへの報告が必要となります。この報告期限は研究の区分、予測可能性、重篤性に応じて変わります。
研究
区分疾病の区分 予測可能性 重篤性 期限 未承認・
適応外疾病等 予測できない 死亡 7日 その他重篤(※1) 15日 非重篤 定期報告 予測できる 死亡 15日 その他重篤(※1) 30日 非重篤 定期報告 既承認 疾病等 予測できない 死亡 15日 その他重篤(※1) 15日 非重篤 定期報告 予測できる 死亡 15日 その他重篤(※1) 定期報告 非重篤 定期報告 臨床研究の実施によるものと疑われる感染症 予測できない 死亡・その他重篤(※1) 15日 非重篤 15日 予測できる 死亡・その他重篤(※1) 15日 非重篤 定期報告 ※1 その他重篤とは死亡以外の死亡の恐れ、障害、障害の恐れ、入院・入院期間の延長、これらに準じて重篤、後世代における先天性の疾病又は異常を指します。
- Q
- CRBへの疾病等報告はどのようなときに必要になりますか?(医療機器・再生医療等製品を使用する試験の場合)
- A
- 医療機器・再生医療等製品を使用する臨床試験では、上記の疾病等の報告に加えて、重篤な有害事象を引き起こすおそれのある不具合についてもCRBへの報告が必要となります。
研究区分 予測可能性 重篤性 期限 医療機器又は再生医療等製品を用いる研究 未知 - 死亡
- 死亡の恐れ
- 障害
- 障害の恐れ
- 入院・入院期間の延長
- 上記に準じて重篤
- 後世代における先天性の疾病
30日 既知
- Q
- 厚生労働省への報告はどのようなときに必要になりますか?
- A
- 未承認・適応外の医薬品等によって発生した疾病等のうち、重篤なものが対象となります。報告期限は死亡・死亡のおそれの場合に7日以内、その他の重篤な場合に15日以内となります。
- Q
- 厚生労働省への報告はどのようにすればいいですか?
- A
- jRCTにアクセスし、以下の手順でシステム上にて報告様式を作成して行います。詳しくは臨床研究等提出・公開システム操作マニュアル【登録者編】をご参照ください。
- jRCTの登録者ページにログインし、「疾病等報告」をクリックする。
- 医薬品の場合には「医薬品の報告」、医療機器の場合には「医療機器の報告」をクリックする。
- 当該疾病等報告の情報を入力する。(一時保存可能)
- 必要に応じて、添付ファイル(PDF)を添付する。
- すべての情報を入力し終えたら、「入力内容確認画面へ進む」をクリックする。
- 入力内容を確認後、「PMDAにメールを送信」をクリックする。
- ステータスが「報告済み」になっていることを確認する。
- Q
- 多施設共同研究で疾病等が発生した場合はどうすればいいですか?
- A
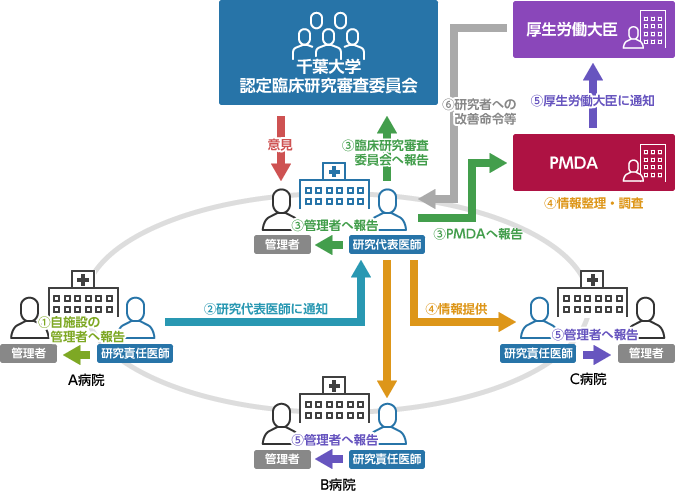
- 当該研究で、自機関のCRBを利用している場合には、自機関のCRBと、必要に応じて厚生労働大臣(PMDA)へご報告ください。CRBで審議結果が出ましたら、その結果を他の実施医療機関に伝えて、各施設の管理者許可を得てください。多施設共同研究で、責任医師として参加している場合には、当該研究の疾病等報告に関する手順に従い、研究代表医師に報告し、他施設のCRBで審議を行ってください。
例:A病院で、特定臨床研究による疾病等が発生した場合
定期報告
- Q
- 定期報告とは何ですか?
- A
- 臨床研究の実施中は、1年毎に定期報告が必要です。具体的な報告内容としては、同意取得例数や実施例数、疾病等や不適合の件数、利益相反の確認となります。詳しくはこちらをご覧ください。
- Q
- 定期報告で報告する有害事象はどのように調べたらいいのですか?
- A
- 千葉大学のデータセンターご利用の場合は、データセンター(hspdatacenter-jim(アットマーク)chiba-u.jp)にお問い合わせください。
- Q
- 定期報告はいつする必要があるのですか?
- A
- 定期報告は、原則として、実施計画がjRCTに初めて公開された日を起点日に、1年ごとに、当該期間満了後2カ月以内にCRBに報告する必要があります。例えば、2022/7/6に公表された試験の場合、2023年度分の報告(2022/7/6~2023/7/5の状況報告)は、当院では遅くとも8月のCRB審議に間に合うように、CRBに定期報告書をCRB事務局 (prc-jim(アットマーク)chiba-u.jp)へご提出ください。
- Q
- 分担医師の変更がある際には定期報告で変更後の分担医師リストを提出するだけでいいですか?
- A
- 定期報告の中で、分担医師の変更はできません。別途変更審査依頼書をご提出ください。
重大な不適合報告
- Q
- 不適合とは何ですか?
- A
- 不適合とは、臨床研究が法令又は研究計画書に適合していない状態を指します。不適合を責任医師が知ったときには、実施医療機関の管理者に統一書式7_重大な不適合報告書で報告することが義務付けられています。
- Q
- どのような不適合が重大になりますか?
- A
- 不適合であって、特に重大なものとは、臨床研究の対象者の人権や安全性及び研究の進捗や結果の信頼性に影響を及ぼすものをいいます。
(例)
選択・除外基準や中止基準、併用禁止療法等の不遵守(ただし、臨床研究の対象者の緊急の危険を回避するためその他医療上やむを得ない理由により研究計画書に従わなかったものについては含みません。)
下記に例示するような場合は、研究の内容にかかわらず、当該重大な不適合に当てはまると考えられます。
- ①説明同意を取得していない場合
- ② 実施医療機関の管理者の許可を取得していない場合
- ③ 認定臨床研究審査委員会の意見を聴いていない場合
- ④ 研究計画からの逸脱によって研究対象者に健康被害が生じた場合
- ⑤ 研究データの改ざん又はねつ造があった場合
- ⑥ その他、認定臨床研究審査委員会が重大な不適合と判断した場合
- (臨床研究法施行通知 規則第15条第3項関係)
記録の保管
- Q
- 責任医師はどのような書類を保管する必要がありますか?
- A
- 以下の文書を、特定臨床研究が終了した日から5年間保管してください。
- 実施計画
- 研究計画書
- 説明・同意文書
- 医薬品等の概要を記載した書類
- 総括報告書
- モニタリングに関する文書(モニタリング手順書など)
- 監査に関する文書(実施した場合に、監査手順書など)
- 原資料等
- 特定臨床研究の実施に係る契約書
- 医薬品等の製造年月日、製造に関する記録
- 医薬品等を入手した数量、年月日の記録
- 医薬品等の処分の記録
- 審査結果通知書
- 実施研究機関における臨床研究の実施許可通知書
- 特定臨床研究を実施するために必要な文書
- 臨床研究法施行規則により研究責任医師が作成した文書、写しまたは記録
終了報告
- Q
- まだ論文がアクセプトされていないので、結果をjRCTに登録したくありません。
- A
- 研究が終了した場合、終了手続きが必要となります。終了の手続きは主要評価項目データの収集期間が終了した日、又は全てのデータの収集期間が終了した日から1年以内に行ってください。総括報告書の概要の公表については、CRBに論文投稿中の旨を報告した上で、公表日を未来日とすることが可能です。また、論文等が公表された場合には、直ちに公表ください。詳しくはこちらをご覧ください。
申請者の方へ
![]()

- 〒260-8677
千葉県千葉市中央区亥鼻1-8-1
TEL: 043-222-7171(代表)
Copyright © 千葉大学 認定臨床研究審査委員会. All Rights Reserved.