千葉大学医学部附属病院 未来開拓センター
〒260-8677 千葉県千葉市中央区亥鼻1-8-1 TEL:043-222-7171(代表)
研究紹介

千葉大学病院
未来開拓センター
千葉大学では家族性レシチン・コレステロールアシルトランスフェラーぜ(LCAT)欠損症を対象とした治療方法の研究が進んでいます。この聞きなれない名前の病気は、コレステロールの代謝に働く酵素(LCAT)が生まれつき機能しないためにおこります。コレステロールの代謝が機能しないと、不要な脂質が不適切に臓器に蓄積するため、いろいろなからだの不調が引き起こされます。それにもかかわらず、現時点では未だこの病気の治療方法がありません。そのため、この病気は難病として指定されています(指定難病259)。
このページではこの病気の特徴を解説し、さらに、千葉大学で取り組まれているこの病気の新しい治療方法の開発について紹介します。
角膜とは、黒目を覆う透明の膜ですが、コレステロールや脂質が蓄積されると徐々に濁ってきます。子供のころから医者から指摘を受けていたり、次第に暗くなると物が見えづらくなり、車の運転が怖くて出来なくなってきた、といった症状を指します。
検診などで継続して蛋白尿の指摘をうけている
HDL-コレステロール値が25 mg/dL未満の方
・現時点で異常がない場合でも、遺伝子異常の種類によっては、将来的に腎機能障害から腎不全を起こすリスクがあります。それを明らかにするためには、精密検査が必要です。
このような臨床初見に合致する、もしくは疑わしい患者さんがおられましたら、遺伝子解析・症状解析等のお手伝いをさせていただきますので、下記までお問い合わせください。
千葉大学では、患者さんの脂肪組織を培養して得られたご自身の脂肪細胞をLCAT酵素を分泌する細胞に作り変え、患者さんに投与することでLCATを持続的に体内で作り出し、症状を長期に改善できるような治療法の開発を進めています。
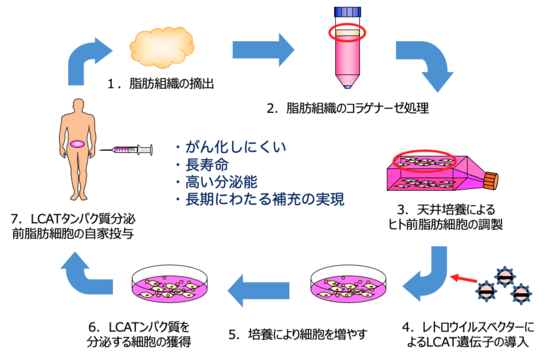
図. 脂肪細胞を用いた家族性LCAT欠損症の治療法
現在、再生医療等安全性確保法下での第一種再生医療臨床研究での成果(Aso M, et al. Heliyon. 2022; 8: e11271.)をうけて、LCAT欠損症の患者さんを対象とした医師主導治験を実施しています。詳細についてお聞きになりたい場合は、以下の連絡先までご連絡ください。
連絡先:千葉大学医学部附属病院未来開拓センター 特任准教授 黒田正幸
E-mail:kurodam(@)faculty.chiba-u.jp